ScanFold Web Server
Welcome!
Using the ScanFold web server you can identify regions within large RNA sequences which generate unusually stable secondary structures (one of the hallmarks of a functional RNA).
The underlying ScanFold algorithm was originally described here (where example uses and verifications are demonstrated): ScanFold: an approach for genome-wide discovery of local RNA structural elements—applications to Zika virus and HIV. Simply input an RNA sequence into the ScanFold (Full Pipeline) server to map its structural landscape and identify uniquely stable RNA secondary structures.
Each of the subroutines comprising ScanFold have also been given separate servers:
- ScanFold-Scan runs a scanning window analysis on an input sequence.
- ScanFold-Fold takes the results of a scanning window analysis (e.g. the output of ScanFold-Scan; see ScanFold User Guide for details on formatting scanning window analysis results from other programs to be used with ScanFold-Fold) and identifies the individual base pairs which contributed most to low z-score windows.
NEW: Run ScanFold 2
Run ScanFold
Visualization
The results of ScanFold are output directly to an embedded igv.js browser as well as downloadable text files. Only the most significant base pairs (as determined by their z-scores) are depicted directly on the browser as arcs, and base pairs which contributed to particularly unusual (potentially functional) structures are colored blue (yellow and green are less favorable, and grey base pairs were from structures no more stable than random). In the example below, we simply input the region of the human VEGFA mRNA three prime UTR known to contain a functional RNA secondary structure: a riboswitch. The blue base pairs (considered most significant) match the previously described model of the riboswtich.
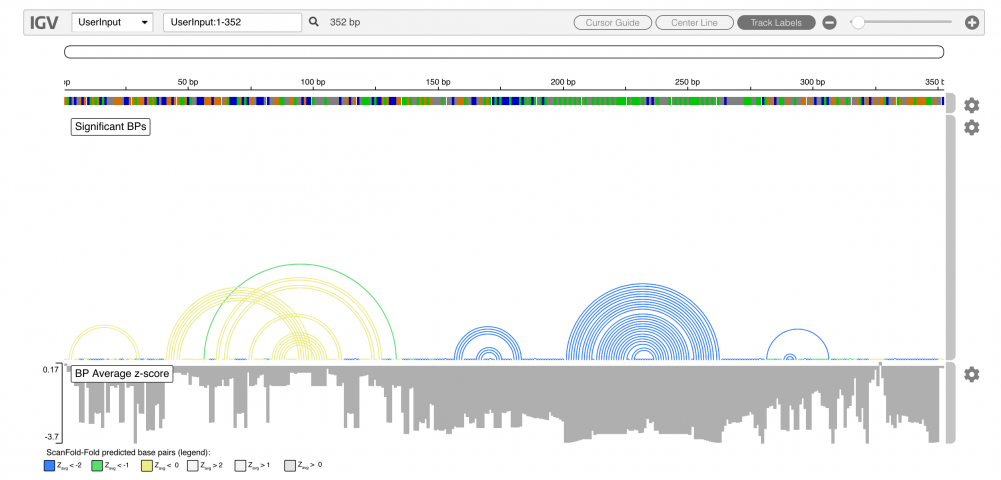
Below each base pair (or unpaired nucleotide) are the average z-scores (Zavg) from the windows in which they appeared. Details about the z-score and other metrics can be found below; briefly however, more negative z-scores suggest a higher likelihood of functionality, and in the example above, highly negative Zavg base pairs, are derived from a known functional RNA. Details about interpreting results can be found in the user guide below.
ScanFold User Guide (PDF)